 |
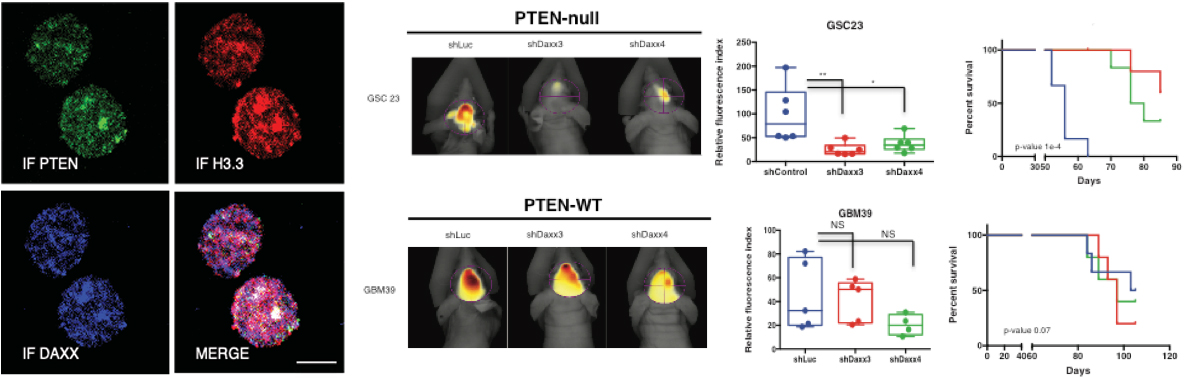
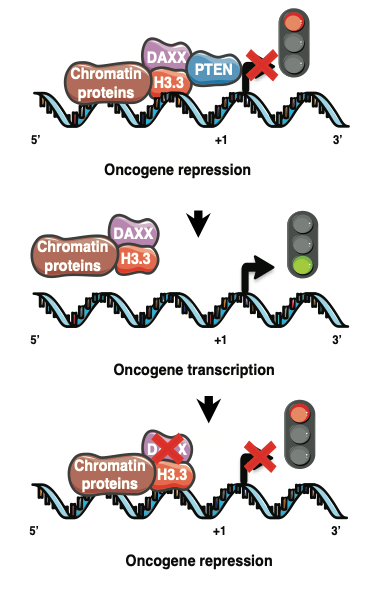
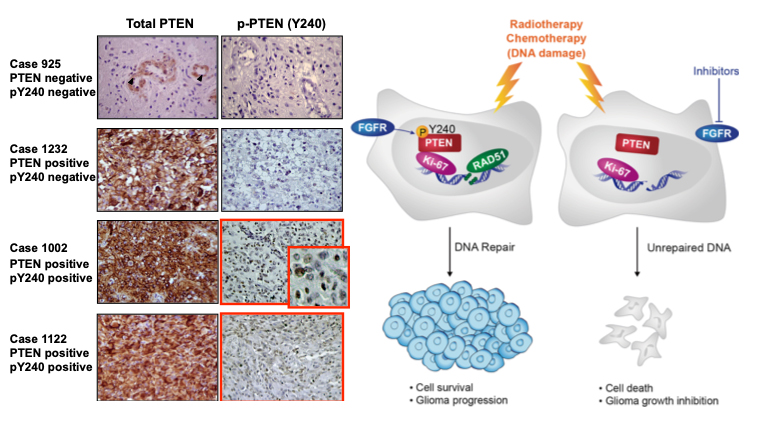
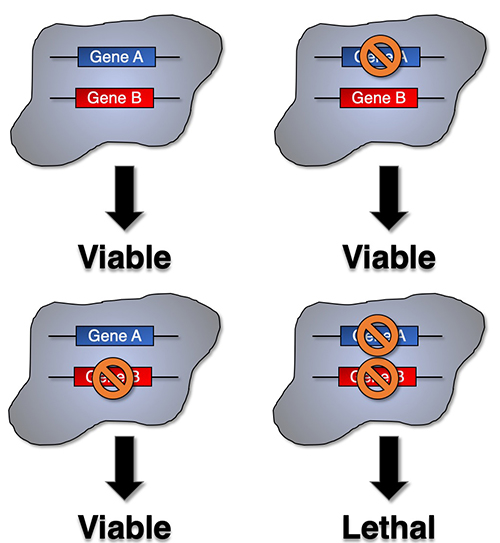
Do cancer cells have Achilles’ heels? The answer may lie in a concept called synthetic lethality, a therapeutic opportunity that arises when a combination of deficiencies in the expression of two or more genes leads to cell death, whereas a deficiency in only one of these genes does not. Using genetically characterized PDX models and glioma neurosphere lines our lab has embarked on synthetic lethal approaches to discover tumor vulnerabilities and genetic dependencies in GBMs harboring alterations of the PTEN tumor suppressor gene. |